- Medizinprodukte
- Beratung & Zulassung
-
-
- EU Marktzulassung
- CE Zeichen Zulassung und Zertifizierung nach MDR
- Technische Dokumentation für Medizinprodukte nach MDR
- Risikomanagement-Akte für Medizinprodukte nach EN 14971
- Grundlegende Sicherheits- und Leistungsanforderungen (GruSuLa)
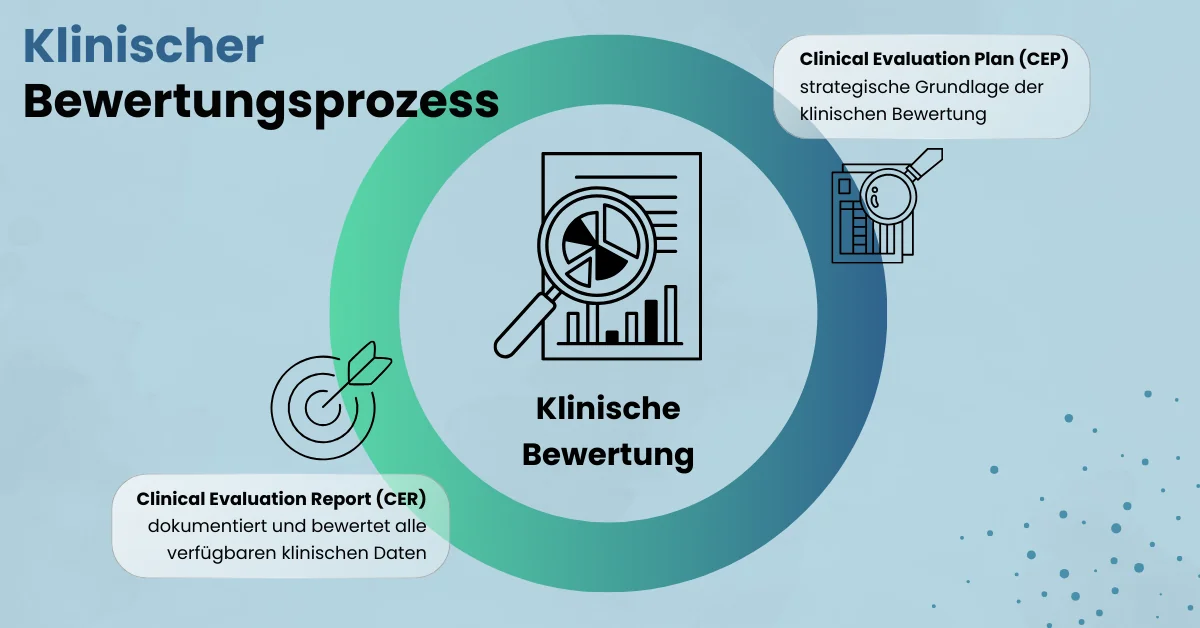
- Klinische Bewertung MEDDEV 2.7/1
- Post-Market-Surveillance (PMS & PMCF)
- Abweichungen in der Technischen Dokumentation nach MDR
- In-Vitro Diagnostika (IVD)
- EU Marktzulassung
-
-
- Weiterbildung
- Über WQS
- Kontakt